
































































































































































































































Nature I 中国科学家提出“药物设计第一性原理”,创新 “脉冲式激活”药物研发新理念,为胆汁酸相关肝病带来新希望
北京时间 2026 年 6 月 10 日,国际顶级学术期刊《自然》(Nature) 在线发表了中国科学院上海药物研究所徐华强研究员团队和李佳研究员团队合作研发的最新研究成果——《一种用于胆汁酸相关肝病的首创脉冲式 FXR 激动剂》(“A first-in-class pulsatile FXR agonist for bile-acid-related liver diseases”)。该研究也是原创新药研究全国重点实验室和生命过程小分子调控全国重点实验室的重要进展。

研究团队系统性提出“药物设计第一性原理”(the first principle of drug design),通过脉冲式激活模拟内源性胆汁酸的生理性动态波动,设计并合成了非胆汁酸类FXR(法尼醇X受体)脉冲式激动剂——原创候选新药Linafexor(CS0159),并完成了从理论创新到临床验证,推进了新药研发底层逻辑的重要变革。
简单来说,“药物设计第一性原理”就是“道法自然”,让药物设计顺应人体的生理节律。当大多数新药都在追求“在体内停留得越久越好”时,研究团队反其道而行之,设计了这款“来去匆匆”的药物Linafexor,让它像潮水、像心跳一样“按一下、松一下”地激活靶点。这款药在动物模型中显示出良好疗效,在 I 期临床中也表现出较好的安全性,为代谢功能障碍相关脂肪性肝炎(MASH)、原发性胆汁性胆管炎(PBC)、原发性硬化性胆管炎(PSC)等胆汁酸相关肝病的治疗,提供了新的设计思路。
这项工作的特别之处在于:它完整呈现了一条从“原始理论创新”走向临床研究阶段的路径——先提出新的药物设计理论,再据此设计出具体分子,并在结构、药代、动物药效以及人体 I 期临床中逐步加以验证。这为基础研究向原创新药的转化提供了一个有价值的研究范例。
目前,Linafexor已获美国FDA授予原发性胆汁性胆管炎(PBC)适应症“突破性疗法认定”(BTD)和“孤儿药资格”(ODD),II期临床研究已完成并取得积极结果,针对PBC的III期注册临床研究已在国内启动,美国PBC III期临床研究正在筹备中。
先读懂三个关键词
在了解这项成果之前,先用三个关键词把背景讲清楚:
关键词 1 · 胆汁酸 由肝脏分泌、储存于胆囊的“天然清洁剂”,帮助人体消化吸收脂肪,同时还像“信使”一样调节全身的糖脂代谢。健康状态下,它会随进餐与空腹“潮起潮落”。
关键词 2 · FXR(法尼醇 X 受体, 也被称胆汁酸受体) 细胞内感知胆汁酸的“总开关”,掌管胆汁酸的合成、转运与清除,是治疗多种肝病的关键药物靶点。
关键词 3 · 脉冲式激活 像心跳一张一弛、潮水有涨有落,身体的信号本是“间歇式”的。本研究主张:好药应顺应这种节律“按一下、松一下”,而不是“一直按住开关不放”。
一、疾病背景:胆汁酸相关肝病的危害与未被满足的治疗需求
这项研究针对的,是一组与胆汁酸代谢异常相关的进展性慢性肝病。它们病因各异,但有一个共同点——胆汁酸代谢失衡、在肝脏内异常蓄积,使肝脏长期受损。
代谢功能障碍相关脂肪性肝炎(MASH),与肥胖、糖尿病、代谢综合征密切相关,患病人群庞大且呈上升趋势。肝脏在脂肪堆积的基础上发生炎症和损伤,若不加干预,可逐步进展为肝纤维化、肝硬化,部分患者可发展为肝癌或肝衰竭,最终引起死亡,是相关肝病的重要病因之一。
原发性胆汁性胆管炎(PBC),是一种慢性自身免疫性肝病:免疫系统“误伤”肝内的小胆管,导致胆汁淤积、胆管被逐渐破坏,多见于中年女性。约 40%–50% 的患者对一线药物熊去氧胆酸(UDCA)反应不足。若控制不佳,疾病可逐步进展为肝硬化、肝衰竭,部分患者最终需要肝移植。若得不到有效治疗,出现症状后的PBC患者的中位生存期仅约 8–10 年,病程迁延、逐步恶化,凶险程度堪比一种"慢性癌症"。
原发性硬化性胆管炎(PSC),以肝内外胆管的慢性炎症和纤维化为特征,胆管逐渐狭窄、瘢痕化,胆汁排出受阻。目前尚无被批准、能明确阻止其进展的药物。PSC 通常进展缓慢,可逐步发展为胆汁性肝硬化和肝衰竭,并增加胆管癌风险,是肝移植的重要病因之一。与 PBC 类似,PSC 病程迁延、逐步恶化;由于至今缺乏有效的疾病修饰治疗,自诊断起至死亡或需肝移植的中位生存期通常仅约 12–18 年,堪称一种进展缓慢的"慢性癌症"。
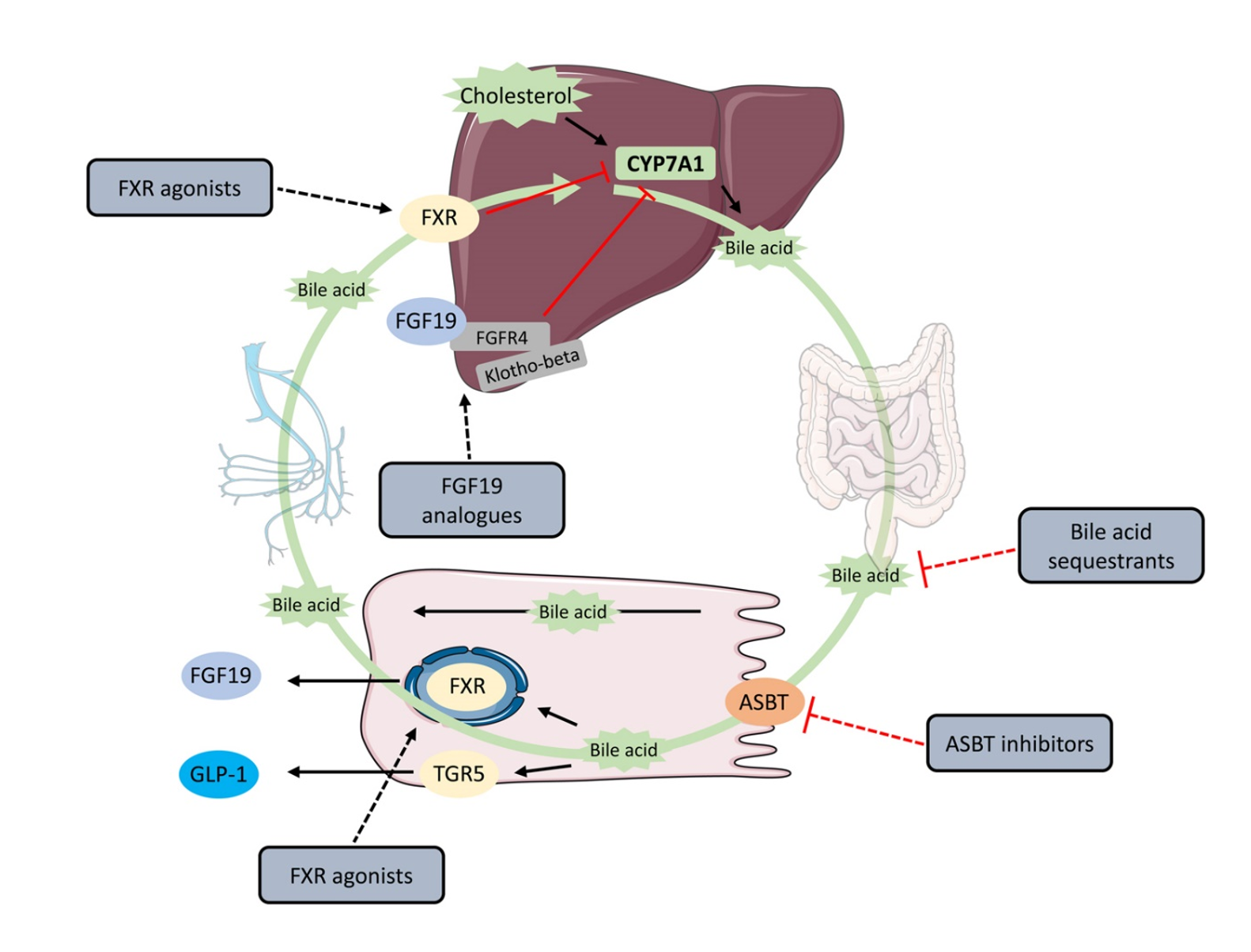
FXR – 胆汁酸代谢的主调控因子
这三种疾病起因不同,却有相似的结局——胆汁酸在肝内异常蓄积、肝脏持续受损。一旦进展到肝硬化或肝衰竭,可用的治疗手段较为有限,肝移植往往是最后的选择,而供体资源紧张。因此,从机制上调节胆汁酸代谢的新药,对应着明确的、尚未被满足的临床需求。
二、长期被忽视的“时间”:为什么过去的 FXR 新药接连失败
新药研发具有复杂性和高风险性——即使进入临床的候选药物中,仍有超过 90% 最终无法获批上市。失败的常见原因,是在可耐受剂量下毒性过大或疗效不足。
长期以来,制药界普遍把“长半衰期、持续高暴露”作为优点,以减少服药次数。然而人体的许多生理过程本质上是节律性、脉冲式的。以胆汁酸为例:进餐后它的水平短暂升高,空腹时又迅速回落,FXR 这个“总开关”也随之一开一合。问题在于,过去20余年进入临床的 20 多个 FXR 激动剂几乎都是“长效”设计——相当于把开关一直按住不放。tropifexor、cilofexor 始终未能获批;奥贝胆酸(OCA)虽于 2016 年获批治疗 PBC,却在欧洲退市、且从未获 FDA 完全批准。或许是这种与身体节律相违背的设计,导致受体逐渐“失灵”,甚至加重病情。
三、回归“第一性原理”:让药物顺应身体的节律
徐华强研究员团队和李佳研究员团队合作提出了药物设计的“第一性原理”:健康是一种动态的生理稳态,疾病是这种稳态被打破,理想的药物应当恢复并强化正常的生理节律,而非将其打破。
基于这一理念,他们“反其道而行之”,刻意设计出一款“快进快出”的分子 Linafexor:它进入体内后会被迅速代谢清除,从而实现与天然胆汁酸波动同步的“脉冲式”FXR 激活——用药后受体被短暂、强力地激活,随后迅速回落、获得休息恢复,避免了“长期疲劳”而失灵。
四、强效精准的“钥匙”:纳摩尔级活性的非胆汁酸小分子
Linafexor 是一款非胆汁酸类小分子 FXR 激动剂。可以把它想象成一把为 FXR “锁孔”量身打造的钥匙——它的激活活性极高,半数有效浓度(EC50,即让一半靶点被激活所需的药物浓度,数值越低越强效)低至 0.35 纳摩尔,与长效药 tropifexor 相当,约为奥贝胆酸的 800 倍。
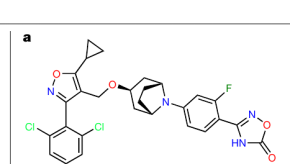
图|Linafexor(CS0159)的化学结构——一把为 FXR 量身设计的“分子钥匙”
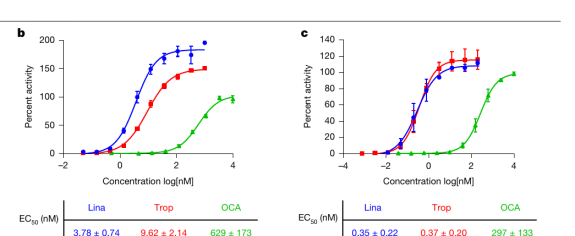
图|在无细胞结合实验(左)与细胞功能实验(右)中,Linafexor(Lina,蓝色)的激活活性与 tropifexor(Trop,红色)相当,远高于奥贝胆酸(OCA,绿色)
五、看清“钥匙如何咬合锁孔”:晶体结构
为研究 Linafexor 为何如此有效,团队解析了它与 FXR 结合的晶体结构(PDB 编号 9LQ3)。所谓晶体结构,就像为“药物分子”和“靶点蛋白”拍下一张原子级别的“合影”。从这张“合影”可以看到:Linafexor 如同一把精心打磨的钥匙,严丝合缝地嵌入 FXR 的“锁孔”(配体结合口袋),通过氢键与疏水作用稳稳“咬合”,把受体锁定在“开启”状态。与已进入临床的 tropifexor 相比,两者在口袋中的结合姿态几乎完全重合(结构偏差仅 0.59 埃),从分子层面印证了 Linafexor 的高效结合。
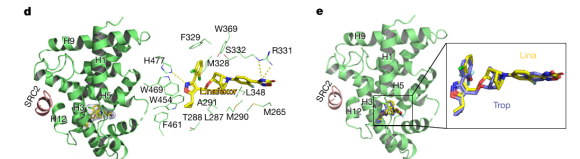
图|Linafexor 与 FXR 结合的晶体结构。左:Linafexor(黄色)嵌入 FXR 受体(绿色)的结合口袋,与多个氨基酸位点形成相互作用;右:Linafexor(黄)与 tropifexor(蓝)在口袋中的结合姿态高度重合
六、“快进快出”的药代动力学:半衰期不到 1 小时
与“长效”老药截然不同,Linafexor 被刻意设计成“快进快出”。这里的关键指标叫半衰期——即药物在体内浓度下降到一半所需的时间,反映它在体内的“停留时长”。在小鼠、大鼠、犬、猴等多个物种中,Linafexor 的半衰期都不到 1 小时,远短于 tropifexor(14–22 小时)和奥贝胆酸(约 24 小时)。这意味着它“用完很快被身体清走”,不会在体内日积月累地堆积。
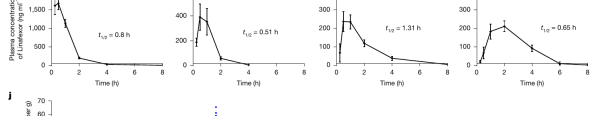
图|Linafexor 在小鼠、大鼠、犬、猴体内的血药浓度随时间变化曲线,各物种半衰期(t₁/₂)均不到 1 小时——快速达峰、快速清除
进一步的分布研究显示,给药后 Linafexor 主要富集在最需要它的部位——肝脏、小肠和胃,并在 24 小时内从所有组织中清除干净,真正实现了与胆汁酸节律同步的“脉冲式”暴露,避免了药物在肝脏蓄积带来的损伤风险。
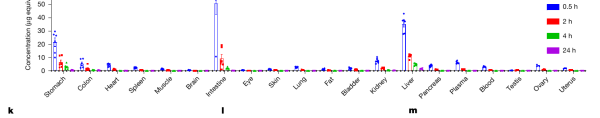
图|Linafexor 在大鼠各组织中的分布:给药 0.5 小时(蓝)主要集中在胃、肠、肝,到 24 小时(紫)已基本清除
七、广谱疗效与一个“决定性实验”
在 MASH、肝纤维化、PBC、PSC 等多种动物疾病模型中,Linafexor 均显著改善了肝损伤、炎症与纤维化指标,疗效优于现有对照药物。而本研究最具说服力的,是一组“同药不同给法”的对照实验:
当研究者改用渗透泵,把完全相同的 Linafexor 分子由“脉冲式”给药改为“持续给药”时,动物出现了严重的全身毒性;而每日一次的脉冲式给药则安全且有效。这一鲜明对比直接证明——决定药物安全与疗效的关键,不是分子结构本身,而是受体被激活的“时长”。团队据此提出“胆汁酸抵抗”概念,与人们熟知的“胰岛素抵抗”相类比:长期过量的信号刺激,会让受体“脱敏”、通路失灵。
八、人体验证(上):药物精准“点亮”靶点
在美国完成的 I 期临床试验(ClinicalTrials.gov 注册号 NCT05082779)中,人体数据与设计预期高度吻合。Linafexor 口服后被迅速吸收、迅速清除,半衰期同样不到 1 小时;血药浓度随剂量成比例增加,而且几乎不受进食影响,给药相当灵活。
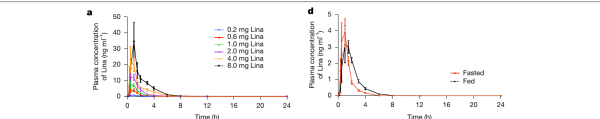
图|人体 I 期临床的药代动力学。左:不同剂量下血药浓度快速达峰、约 8 小时内清除;右:进食(Fed)与空腹(Fasted)对药物影响很小
更重要的是,药物精准“点亮”了靶点:反映 FXR 被激活的标志物 FGF19 明显升高,反映胆汁酸合成的标志物 C4 显著下降,二者都在 24 小时内回到基线水平——在人体中再现了“脉冲式”激活的特征。
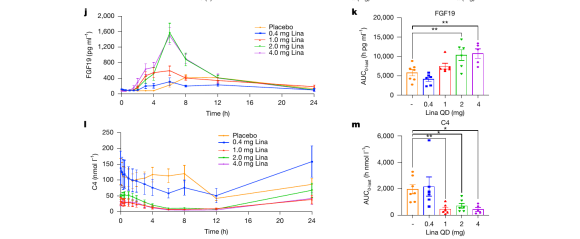
图|人体 I 期临床中,Linafexor 剂量依赖性地升高 FGF19(上,靶点被激活)、抑制 C4(下,胆汁酸合成下降),并均在 24 小时内回归基线
九、人体验证(下):零药物相关不良事件
安全性是 FXR 类药物历史上的“老大难”。而在本次 I 期临床的所有单次与多次递增剂量队列中,Linafexor 组均未出现与药物相关的不良事件——这与既往一些因长半衰期而常伴随明显副作用的 FXR 激动剂形成对照,为其后续临床开发提供了支持。
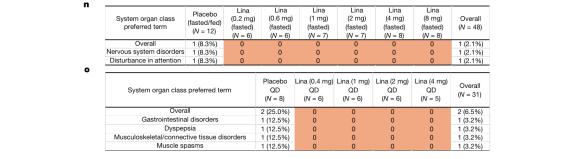
图|单次(上)与多次(下)递增剂量队列的安全性汇总:Linafexor 各剂量组(橙色高亮)药物相关不良事件均为 0
在 I 期临床良好表现的基础上,Linafexor(CS0159)已在原发性胆汁性胆管炎(PBC)与代谢功能障碍相关脂肪性肝炎(MASH)两项 II 期临床研究中取得积极结果:初步数据显示,该药在这两个适应症中均展现出明确的疗效,并具有突出的安全性表现。两项 II 期临床研究的详细结果将另行专文发表。
十、意义:从原始理论创新到临床研究的重要进展
回到最初的主线——这项研究的意义,在于它走出了一条从原始理论创新到临床研究的扎实路径:一个新的药物设计理论(“第一性原理”与脉冲式激活),通过 Linafexor 在结构、药代、动物疗效及人体 I 期临床中得到了较为系统的验证;该药已完成 II 期、并启动 III 期临床研究,更充分的疗效与安全性结论仍有待 III 期及后续研究确认。这为“从原创理论到原创候选新药”的研发路径提供了一个有价值的范例。
这一思路或具有更广的参考价值。许多生理过程——如激素信号、免疫调节、昼夜节律代谢——同样依赖“节律性”而非“持续性”的信号。在药物设计中考虑这些天然节律,可能有助于降低部分新药因毒性或治疗窗口过窄导致的研发风险。
研究团队与合作单位
本研究主要由中国科学院上海药物研究所徐华强研究员团队和李佳研究员团队合作完成,合作单位还包括凯思凯迪(上海)医药科技有限公司、上海交通大学医学院附属瑞金医院、厦门大学、临港实验室等。徐华强研究员长期从事 G 蛋白偶联受体(GPCR)与核受体的结构生物学及相关药物研究。李佳研究员长期从事代谢性疾病机制、靶标发现和创新药物研究。Linafexor(CS0159)由徐华强研究员团队和李佳研究员团队合作发现,由上海药物所与美国温安洛研究院(Van Andel Research Institute)以独占许可方式转化至凯思凯迪(上海)医药科技有限公司,开展后续研发。
论文信息
论文标题(英文):A first-in-class pulsatile FXR agonist for bile-acid-related liver diseases
中文译名:一种用于胆汁酸相关肝病的首创脉冲式 FXR 激动剂
发表期刊:《自然》(Nature)
DOI:10.1038/s41586-026-10633-1
通讯作者:李佳(jli@simm.ac.cn)、徐华强(eric.xu@simm.ac.cn)
结构数据:FXR/Linafexor 复合物晶体结构已存入 PDB(编号 9LQ3)
临床注册:ClinicalTrials.gov,NCT05082779
(供稿部门:徐华强课题组、李佳课题组;审核:刁文桐;责编:王肖成)